By Huri Mücahit The Food and Drug Administration (FDA), as the name suggests, is the primary regulatory organization for food and drug safety, including biologics and medical devices. However, surprisingly, the FDA regulates much more in the name of protecting public health, such as cosmetics, veterinary products, and tobacco products. The range in regulatory jurisdiction speaks to the long history of food and drug regulation that came about in response to the highly unregulated nature of medicine production in the early 1900’s, resulting in the death of 22 children due to contaminated vaccines. Since then, several laws have been passed requiring the licensing and inspection of food and drug manufacturers, as well as mandating the demonstration of not only safety, but also efficacy of a drug. Peter Marks, M.D., Ph.D., Director for Biologics and Research Evaluation, discussed the FDA’s history and approval process in this iJOBS seminar. Of particular interest to Ph.D. students in the health sciences, is the FDA’s role in promoting the development of products that address the public’s unmet medical needs. The agency addresses these needs through several factors, such as extracting user fees for each application examined, so that performance metrics can be placed on the FDA to ensure timely review. In addition, to further facilitate drug and biologics development, sponsors of the applications, which are typically pharmaceutical companies, can ask for OrphanDesignation, apply for Priority Review vouchers, or apply through any of the expedited development programs. As the first category suggests, the Orphan Designation covers treatments for rare diseases affecting less than 200,000 people, and it features tax credits, 7 years of market exclusivity, and user fee exemption. Priority Review vouchers can be applied for neglected diseases of the tropics, rare pediatric diseases, and for medical countermeasures. This option ensures the review process will be completed within 6 months rather than the standard 10, however, the sponsors must demonstrate significant improvement in safety or effectiveness. Additional programs targeting treatments for serious conditions, like Fast Track, Accelerated Approval, or Breakthrough Therapy, may offer advantages such asrolling reviews in which the committee will review components of the application as they are prepared, approval based on surrogate endpoints, or extensive guidance from the review committee. Finally, sponsors can also be granted the Regenerative Medicine Advanced Therapy Designation (RMAT), if they provide cell therapies, tissue engineering products, or human cell and tissue products. While the FDA has many paths to approval for new treatment applications, the agency naturally follows a standard process to ensure safety and efficacy of the treatment. This might include an initial information meeting between the FDA and the sponsor to go over the application procedure and provide guidance on the types of studies required prior to clinical trials. If the results look promising once the necessary pre-clinical trials are conducted, a manufacturing process will be developed, keeping with Good Manufacturing Practices. A second meeting might then be scheduled to propose Phase I trials and protocols, which, if approved, will be used to generate data for further review. Upon proving that the treatment has the potential to address an unmet need, the FDA will assign a specific designation, such as RMAT or Fast Track, and review the additional data produced from Phase II and III trials, as well, as manufacturing protocols. Finally, after a series of informal, mid-cycle, and late-cycle meetings, an advisory committee consisting of experts within the field will meet to grant or deny approval. This committee may also require post-marketing studies to be conducted to further test the safety of the treatment. If the sponsor fails to complete these studies, the FDA has the authority to rescind approval. 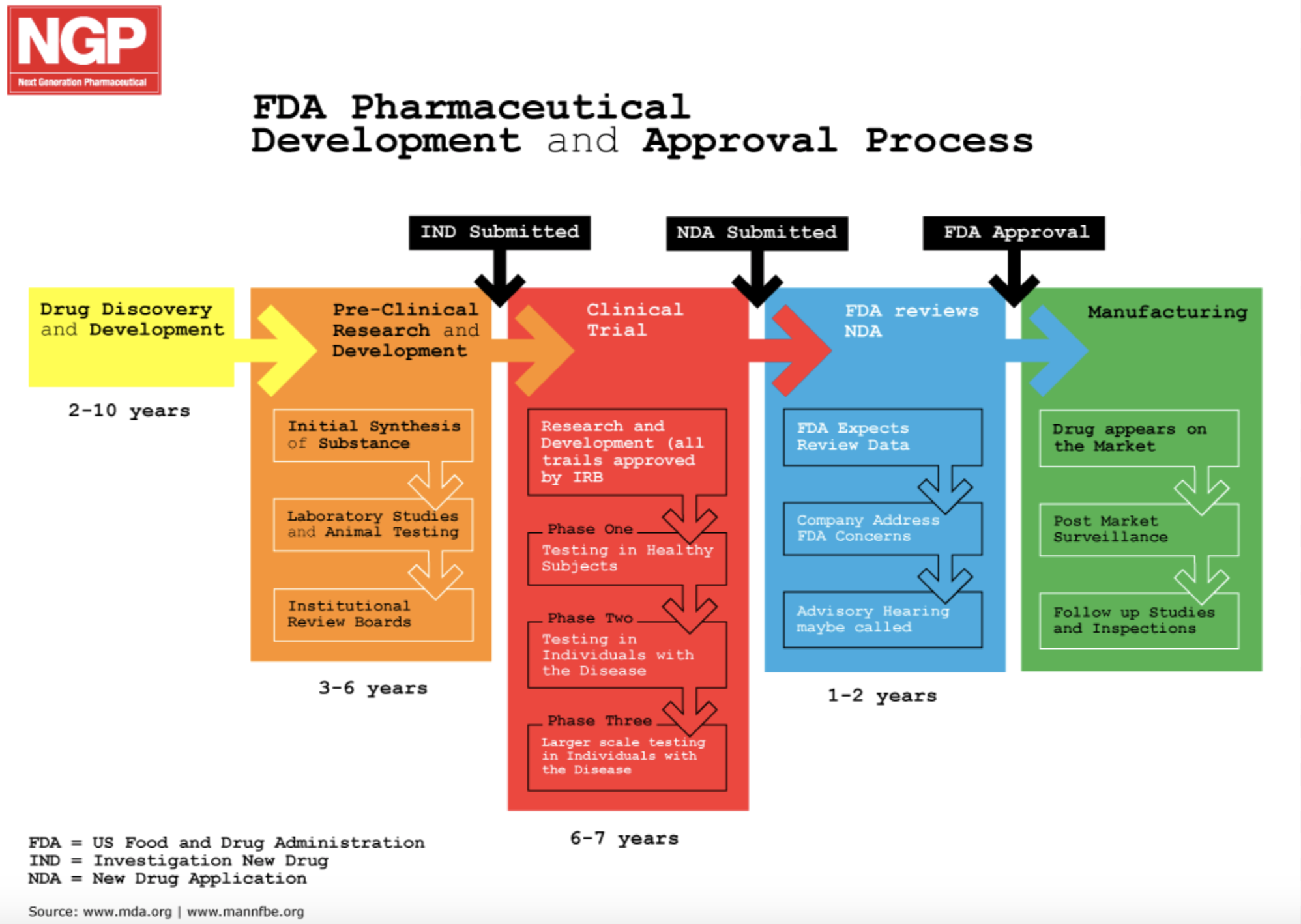 For Ph.D. students interested in working with the FDA, those within epidemiology or biostatistics fields have the highest chance for employment immediately following their defense. However, to be a hired as a regulatory reviewer or research reviewer, post-doctoral research associates are preferred. Additionally, since the laboratories and the majority of offices are housed in the main facility in Silver Spring, Maryland, these positions are only available at this site. If the applicant wishes to remain local, there are inspector positions available throughout the country. The FDA also provides internship opportunities for interested students from a variety of backgrounds, including undergraduates and post-docs. Overall, the FDA is a crucial agency in aiding the development of drugs and biologics and ensuring safety and efficacy of these treatments. Given the sheer number of drug applications received, Ph.Ds. have a wealth of opportunities for employment in reviewing these applications or conducting lab work within the FDA. Ultimately, these opportunities provide a medium to enact significant change and guide the path for new treatments. Edited by: Jennifer Casiano-Matos and Monal Mehta This blog post was written after attending the iJOBS Career Seminar: Jobs at the FDA on June 13th, 2019.
For Ph.D. students interested in working with the FDA, those within epidemiology or biostatistics fields have the highest chance for employment immediately following their defense. However, to be a hired as a regulatory reviewer or research reviewer, post-doctoral research associates are preferred. Additionally, since the laboratories and the majority of offices are housed in the main facility in Silver Spring, Maryland, these positions are only available at this site. If the applicant wishes to remain local, there are inspector positions available throughout the country. The FDA also provides internship opportunities for interested students from a variety of backgrounds, including undergraduates and post-docs. Overall, the FDA is a crucial agency in aiding the development of drugs and biologics and ensuring safety and efficacy of these treatments. Given the sheer number of drug applications received, Ph.Ds. have a wealth of opportunities for employment in reviewing these applications or conducting lab work within the FDA. Ultimately, these opportunities provide a medium to enact significant change and guide the path for new treatments. Edited by: Jennifer Casiano-Matos and Monal Mehta This blog post was written after attending the iJOBS Career Seminar: Jobs at the FDA on June 13th, 2019.
iJOBS Blog