By Natalie Losada
Image of script for processing genomics data, with images of DNA (large circle) and cells under a microscope (small circle).
Computing and bench work might feel like two different worlds. But much like other dualisms—sun and moon, sound and silence—they are complementary, and each necessary. By uniting both computational and experimental research, you can make your research more impactful. Most of us are already skilled at the bench, so the Data Carpentry community sought out to fill in the gaps of basic computing skills for researchers.
The Genomics Data Carpentry workshop was held from January 26-29, 2021, and taught by instructors (this link has a full list of instructors’ Twitters, ORCIDs, and GitHub links!) and helpers from all over the globe. It included researchers from Italy (Monah Abou Alezz – postdoctoral fellow, San Raffaele Hospital), Poland (Aleksander Jankowski – Assistant Professor, University of Warsaw), and the United Kingdom (Vasilis Lenis – lecturer, Teesside University, and Nadine Bestard – University of Edinburgh). The Data Carpentry community was phenomenal at explaining and documenting every lesson, so I recommend you take a look at their website to learn at your own pace.
Screenshot of workshop – slide from Monah Abou Alezz’s presentation. Featuring Janet Alder in top-right, Monah just below, and other instructors and helps for the workshop.
The workshop was predominantly a hands-on experience. But rightfully so, it started out with a word to the wise. If you take nothing else from this article, take away this lesson: the importance of pre-project organization and management.
"Think like computers when you are using spreadsheets: they cannot separate your arbitrary sample names like you can."
- Monah Abou Alezz
Organizing data can easily be discouraging for people, but it doesn’t have to be if we think about it strategically before generating any data. Your metadata should come first, which is essentially “the data about the data”. For example, your DNA sequences are data, but your metadata includes what organism it comes from, the particular strain, any known mutants, sample concentration, and much more. Researchers normally keep a written lab notebook with experimental details, but Monah insisted on keeping an electronic spreadsheet (Microsoft Excel, LibreOffice Calc, Google Sheets) with metadata and data for all experiments. He also emphasized “data tidiness”. If you want to automate data analysis, you need to have spreadsheets that are easy for computers to understand. It helps to keep in mind that computers are only able to detect the presence or absence of something. A sample called “green_monster” is not the same as “green_MONSTER” because the computer is detecting the presence or absence of capital letters. For all spreadsheet preparation, you should follow these notes:
- Avoid spaces in column, row, and sample names
- Use underscores and dashes. In coding, spaces are required for some commands to separate the action from the sample. If the sample name has two words, the computer will only identify the first.
- Keep single types of information in each column
- If a column has numerical and alphabetic entries, or just if the column seems crowded, do not be afraid to add more columns. This will keep all sample information neatly organized and easily interpretable by the computer.
- Use consistent formatting
- Capitalization, format of dates, decimal accuracy, abbreviations, sequential labeling, all need to be the same. So, decide on your favorite format before filling the spreadsheet and stick with it.
- ALWAYS keep the RAW data untouched
- Copy data to a separate folder where you can work with it (i.e., a “working directory”). If you find something wrong after processing the data, or decide to try methods of analysis, you can still go back to the original, untouched file.
- Stick with CSV and TSV formats
- CSV and TSV, Comma and Tab Separated Values, respectively, are text files that can be read by programming software, while Xlsx files cannot. CSV/TSV files are also smaller and therefore better when working with copious volumes of sequencing data.
Other important considerations (when automating your data analysis)
If you plan on doing Next-Generation Sequencing (NGS) projects for whole genomes of multiple organisms or samples, you might run into some challenges away from the bench. Sequencing files can be quite large. They usually include the genome and quality of each sequence read, and sometimes they are “paired” and contain overlapping reads of the same segment. Therefore, storing raw and working data separately might be cumbersome on a personal computer with limited storage. Ask yourself, can your computer handle the work? Or do you need a cloud service? As a Rutgers student, you have access to Box for storage. Resources like Dropbox or Google Drive might not have enough storage at an affordable rate for all your data, so look around to find appropriate solutions for your projects! You also need to confirm that your files can be accessed by other lab members and that they are in a redundantly backed up location. The recommendation is to back up data thrice: once in a cloud (like Amazon S3, Microsoft Azure, Google Cloud) and on two hard drives in geographically separated locations (like your lab hard drive and campus data storage facility).
Ask yourself, can [your computer] handle the work? Or do you need a cloud service?
Once you know where your data will be stored, you need to plan where it will be processed. Again, can your computer handle it? Or will you need the aid of a high-performance computing center like the Amarel Cluster, which is also available for Rutgers students. The Office of Advanced Research Computing (OARC) are extremely helpful in getting you set up and very responsive should you have any questions while processing your data.
You should also plan ahead to know what programs you will use to process your data. Some software are written in particular languages and can only read certain formats of files. You need a plan for the proper transferring and copying of data to avoid small errors. When you receive your sequencing results, they will be in compressed/zipped files, so unzipping before transferring could result in lost or altered files. If copied with the incorrect software, the format of files could be unintentionally changed and rendered unreadable by some programs. To prevent errors, ensure your samples are properly labelled and logically organized. As mentioned in the previous section, keep all formatting consistent. Label your samples in sequential order so it’s easy for everyone involved, watch out for repeat sample names or barcodes, and make sure you have enough, but not too many columns in the spreadsheet you send for the NGS order. This spreadsheet is different from your own records with the detailed metadata and data, which you will acquire and input after the sequencing results are returned to you.
Once your experiments are completed and ready to be published, you will be working with publicly accessible databases. Two common databases are SRA (Sequence Read Archive) from NCBI (National Center for Biotechnology Information) and ENA (European Nucleotide Archive) from the EBI (European Bioinformatics Institute). You need your raw data to be easily accessible and manageable when transferring to these archives. And, if you’re ever looking for raw data from a particular organism or project, much like searching scientific literature or reading news in this day and age, check all sources. Some places might have information that others don’t!
Beginning to code
The majority of the work involved with computing is learning the computer language for the programs you want to use. An article about everything I learned in this workshop would end up as a long grocery list of commands and notations. With that in mind, I will only discuss the basics needed to start automating your data analysis and some tips to help you learn more quickly.
First, if you’re new to genomics data carpentry, you might want to work with some practice data. We obtained our practice data from NCBI. Our practice data came from a project named Bioproject, which you can search for on the NCBI website with the accession number PRJNA294072. We clicked on the “Long-Term Evolution Experiment with E. coli” in the search results, and then clicked on the total number of SRA experiments. This brought us to a huge list of sub-projects (from here you can click “send results to Run Selector” so they are easier to look through). For more detailed instructions on how to download the data, please check out Data Carpentry’s website. Some databases -such as SRA- require a toolkit to download data from, but others -such as ENA- do not.
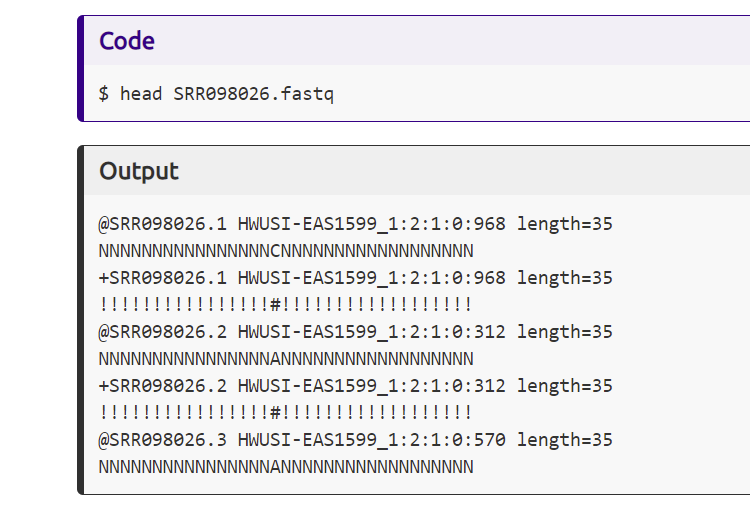
Screenshot from the Data Carpentry website showing the input command to view the top few lines of a sequencing results file. Below the command shows the output which includes metadata in the first line, the sequence in the second line, metadata again in the third line, and the quality of the read using symbol representations.
To actually begin coding and communicating with the computer, you need two things: an interface that allows communication with the computer, called the command line interface (CLI), and a secure way to transfer large files. If you have a Linux or Mac operating system, you’ll have a command line ready to use, called the terminal. If you are a PC user, you will need to download the software PuTTY, which is a terminal emulator. As a Linux or Mac user, you should be able to transfer files from a remote server to your computer, and vice versa, using the command ssh (secure shell), which is a cryptographic network protocol. For PC users, you can download WinSCP (Windows Secure Copy Protocol), to securely transfer files between your computer and a remote server. Alternatively, a PC user could download MobaXTerm that performs the functions of both PuTTY and WinSCP, but you should ultimately choose the software whose interface you understand better.
Tips for learning:
- Spaces are not the absence of information
- Spaces are purposefully placed in commands to separate “arguments” (a math term for a piece of information). A space means “okay, I am done naming my file, now here is the next command/piece of information”.
- Look at what the letters mean
- Example: sometimes the command for a certain software will include a “-o” followed by an instruction to give the final output name. You can easily remember this as “-o” stands for “output file name”.
- Name files and folders accurately
- Think of this like chess, you need to plan five steps ahead when naming. You can’t just name folders “data”, “backup”, “extra”. If you have to redo everything and you have new data and new backup files, how will you differentiate which is which? Be descriptive, but concise.
Keep in mind that it will take a few tries to get your workflow to be efficient. The irony of automating data analysis to save time is that you spend more time in the beginning trouble shooting before you can actually reap the time saving benefits. Also keep in mind that at least half, likely more, of the time spent coding is actually spent googling. The Data Carpentry instructors all agreed on this point, because you will need to spend time learning what programs you can use to analyze genomics data, understanding how the commands work, finding how to connect commands into a single script, and discovering where you usually make your syntax mistakes (of which there will be plenty, it happens to everyone). Ultimately, the best way to learn to code for data analysis is having a problem and trying to solve it. If you don’t already have a problem, find one online and start computing!
This article was edited by Senior Editor Helena Mello and Senior Editor Samantha Avina.