By Nowrin Ahmed, PhD

Are you a Ph.D. student or postdoctoral researcher interested in pharmaceutical sciences (pharma) or biotechnology (biotech)? Are you eager to explore some of the subspecialties within the pharmaceutical industry? If so, you're in the right place! Earlier this year, iJOBS invited Dr. Daniel Gordin to help familiarize potential new hires in the pharma/biotech sector with the concept of pharmacokinetics (PK) and pharmacodynamics (PD). Dr. Gordin has a Ph.D. in Pharmaceutical Sciences with a specialization in pharmacokinetics. He served as a pharmacokinetic reviewer within the Division of Biopharmaceutics at the Food and Drug Administration (FDA). Following his tenure at the FDA, he spent over three decades in regulatory affairs at various pharmaceutical companies, including Novartis.
This blog post delves into the intricacies of PK and PD, as presented by Dr. Gordin, and explains how PK/PD studies play a critical role in the drug development process.
PK is derived from the ancient Greek word ‘pharmakon’ (meaning ‘drug’) and ‘kinetikos’ (meaning ‘moving’). Thus, PK is a branch of pharmacology that studies how drugs move through the body which are tracked through changes in drug concentration in the body over time. It is influenced by processes such as the rate of absorption, distribution, and excretion. In humans, after a drug is swallowed, it dissolves in the stomach and moves to the first part of the small intestine, the duodenum, where it is absorbed into the bloodstream. Drug absorption depends on factors such as dissolution in the stomach, solubility, permeability, and gastric emptying—the process by which food moves from the stomach to duodenum. After absorption, the drug is transported via the blood to the liver, where it undergoes metabolism. The resulting drug metabolites, along with the unchanged drug, are distributed within the body and eventually excreted in urine or feces.
PD is the study of a drug’s effect on the body. This is evaluated in terms of drug maximum effect (Emax) and the concentration that produces 50% of the maximum effect (EC50), as well as the drug’s mechanism of action. Drugs exert their effects by binding to receptors in the body. There are four main types of receptors:
- G-protein-coupled receptors
- Ligand-gated ion channels
- Intracellular receptors
- Tyrosine kinase-coupled receptors
The mechanism of action of a drug is dependent on its interaction with a specific receptor. Drugs that bind to a receptor to produce a biological response are called agonists whereas drugs that bind to receptors to block or reverse the effects of agonists are called antagonists.
How do pharma companies assess different PK/PD parameters?
The strength of the drug, referred to as its effect size, depends on the drug’s binding affinity (Ki) for a receptor. The Ki is the concentration of the drug at which 50% of the receptors are occupied. A lower Ki indicates higher potency. When multiple drugs with different Ki values are present in the bloodstream or tissues simultaneously, drugs with a lower Ki can displace those with a higher Ki, reducing the effectiveness of the latter.
In vitro dissolution testing is performed by the Chemistry, Manufacturing, and Controls (CMC) department of a pharmaceutical company. The CMC is responsible for assessing how well the drug dissolves in the gastrointestinal (GI) tract to determine its bioavailability, the ability of the drug to be absorbed and used by the body, dosage forms, and stability. In vitro dissolution is tested using a dissolution apparatus. The PK team determines the criteria for a single point dissolution test, and it serves as a quality control measurement which must be performed for each batch of drug.
How do routes of drug administration impact PK?
Drugs are administered orally, intravenously (IV), subcutaneously (SC), intramuscularly (IM) and through inhalation. IV is the fastest route of administration because the drug is injected directly into the bloodstream. Each route of administration affects two key PK parameters: maximum concentration (Cmax) and elimination rate (Ke). For example, after IV administration, the drug rapidly reaches its Cmax and is eliminated from the body faster relative to oral administrations, where Cmax is reached gradually.
How is PK/PD established in clinical studies?
Understanding the PK/PD profile of the drug in humans is essential to determine factors such as dosage, efficacy, safety, and formulation of the drug. Drug development begins with preclinical studies in animals or cells and, if the results are promising, transitions to clinical trials in human subjects. Clinical trials are conducted in three phases to ensure patient safety and drug efficacy. Phase I trials, which involve healthy subjects, are conducted to assess drug safety. In this phase, PK studies are performed to examine the drug's profile in a single-dose study. Blood concentrations of the drug are measured over time using high-performance liquid chromatography-mass spectrometry (HPLC-MS).
Key PK parameters include the total drug exposure over time (area under the curve, or AUC), the maximum blood concentration (Cmax), time at which the drug is at half its maximum concentration (t1/2), and the elimination rate (Ke). The half-life is important for determining the steady-state concentration of the drug, which occurs when the rate of administration equals the rate of elimination.
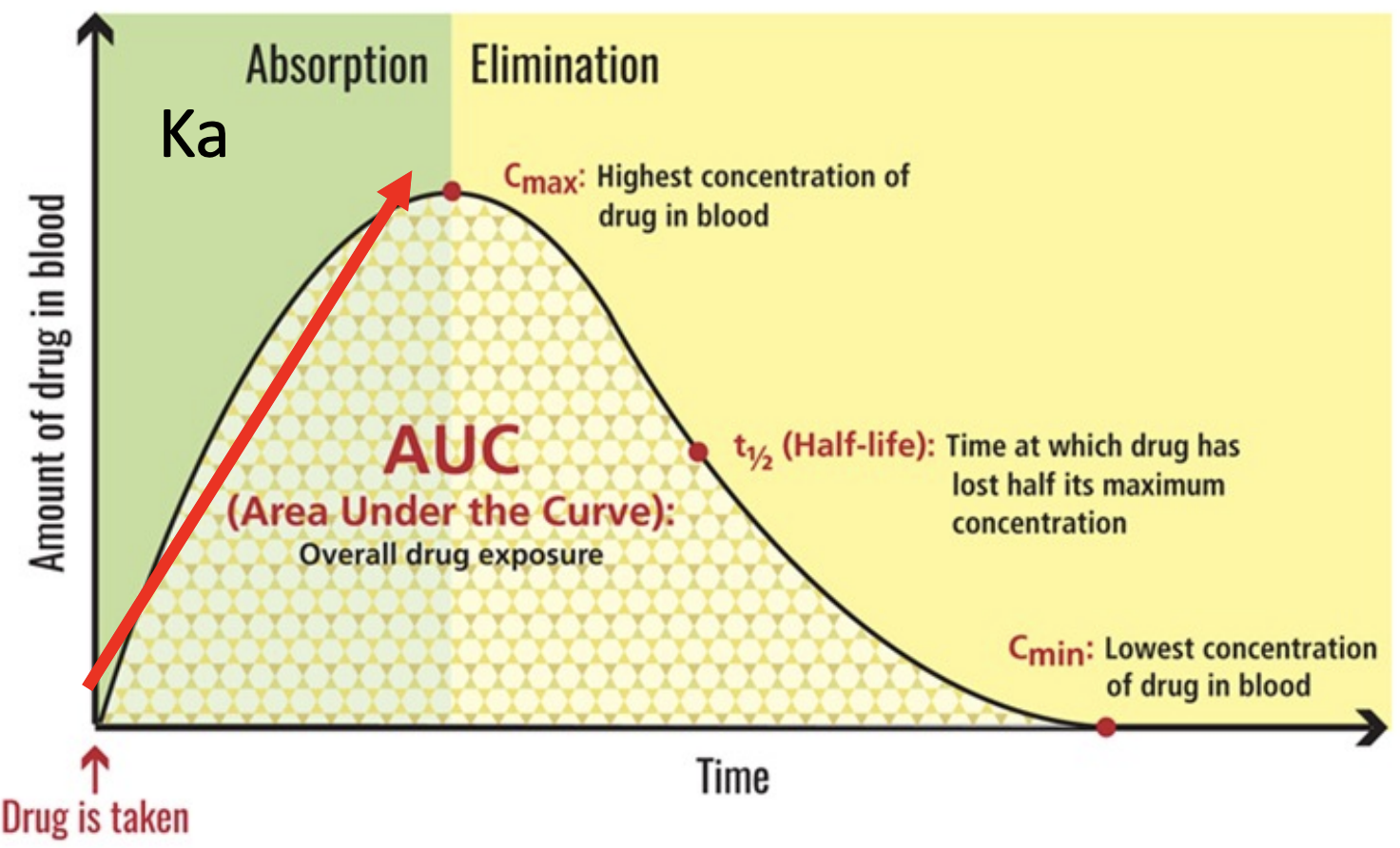
The drug concentration needs to be high enough for the drug to be efficacious and safe to use. Achieving this delicate balance is a major challenge in drug discovery. Therefore, PK studies are a crucial part of the drug development process as dosage and formulation of the drug is determined based on PK studies.
Types of PK studies and their objectives
Placebo controlled PK studies are often conducted in small groups to investigate various properties of new drugs. The following studies were discussed:
|
PK Study |
Objectives |
|
Single ascending dose |
To examine the maximum tolerated dose and dose proportionality and safety |
|
Multiple dose study |
Examines dose linearity, optimum frequency of dosing based on T ½ and safety |
|
Food effect study |
Examines how food content such as high fat food affects drug absorption and safety |
|
Absolute bioavailability study |
Compares drug absorption rate of oral dosage with other form of drug such as in solution |
|
Hepatic impaired study |
Examines how the PK and safety of the drug is influenced by hepatic malfunction |
|
In vitro hepatic enzyme study |
Assesses the hepatic enzymes that metabolizes the drug |
|
Renal impaired study |
Examines how the PK and safety of the drug is influenced by renal malfunction |
|
Population PK modeling |
Assesses how variability such as age, gender, disease progression in patients impacts PK parameters |
|
PK drug-drug interaction study |
Examines how the PK of a drug changes when administered with another drug |
In summary, PK/PD studies are used to guide the selection, optimization, and evaluation of new drug candidates to ensure their safety and efficacy in clinical use. If you are interested in learning more about PK/PD, use the link Dr. Gordin provided here.
This article was edited by Senior Editors Joycelyn Radeny and Antonia Kaz.