By Megan Schupp
In today’s rapidly evolving biomedical landscape, researchers often fall into two categories: bench-focused and computational-focused. However, with the increasing reliance on data science to unlock the complexities of biology, the lines between these fields are blurring. To remain at the forefront of scientific discovery, scientists must not only grasp fundamental biological concepts but also develop computational skills to analyze vast, complex datasets. The future of biomedical research hinges on this integration—bioinformatics.
Recognizing this need, the Rutgers iJOBS program recently hosted a two-day genomics workshop from March 17th to March 18th, facilitated by Data Carpentry. The workshop aimed to provide budding researchers with hands-on experience in genomics and bioinformatics. As my research involves large datasets, including transcriptomics and proteomics, I found the workshop invaluable in equipping me with the essential tools to become a competent bioinformatician.
The Importance of Project Management and Data Organization
One of the key lessons emphasized during the first day of the workshop was the significance of project management and data organization. Without proper planning, computational experiments can quickly become unmanageable. The first step in any bioinformatics project is organizing metadata – “data about the data”. Key details in metadata include organism species, genotype status, sample concentration, and unique identifiers, which facilitate long-term tracking. Metadata standards vary by research field but storing this information in structured formats such as electronic spreadsheets or text files is essential.
Additionally, managing data size is crucial. Next Generation Sequencing (NGS) experiments often produce large file types such as FastQ or BAM files, which can range from 1-2 GB per sample. Given these storage demands, researchers should utilize external hard drives or cloud-based storage solutions. Rutgers offers several options for long-term data storage, including Google Drive (30 GB), OneDrive (5 TB) and Dropbox (5 TB). A best practice for data security is maintaining two copies—one on a cloud-based storage system and another on an external hard drive.
Best Practices for Data Structure and Storage
To ensure efficient data handling, the following guidelines should be observed:
Data Structure
- Preserve raw data – never modify the original files.
- Put all your independent variables, such as strain or DNA-concentration, in columns.
- Use clear column names without spaces; opt for hyphens (-), underscores (_) or camel case.
- Example: ‘library-prep-method’ or ‘LibraryPrep’ is preferable to ‘library preparation method’ or ‘prep’. Keep in mind that computers interpret spaces in particular ways.
- Store variables in separate columns to enhance data usability.
- Example: Instead of E. coli K12 in one column, use separate columns for species (E. coli) and strain (K12). Depending on the type of analysis you want to do, you may even separate the genus and species names into distinct columns.
- Export cleaned data in a universal format like CSV (comma-separated values). This is required by most data repositories and ensures that anyone can use the data.
Data Storage
- Ensure accessibility – both you and other members of your lab should have access.
- Backup data in two locations – preferably in different physical locations.
- Handle data transfers carefully – different file types require different software to access the information. Sequencing results typically arrive in compressed (zipped) files. Be cautious, unzipping before transferring could lead to lost or altered files. Using incorrect software can corrupt file formats, making them unreadable.
- Use proper file labeling – to minimize errors, samples should always be well-labeled and organized.
- Deposit final dataset in public repositories such as Gene Expression Omnibus (GEO) at the National Center for Biotechnology Information. Gene Expression Omnibus (GEO) is a database supported by the National Center for Biotechnology Information (NCBI) that can hold raw and processed data. Many publications require data to be deposited into public repositories before publishing can occur.
Bioinformatics Workflow for RNA-Seq Analysis
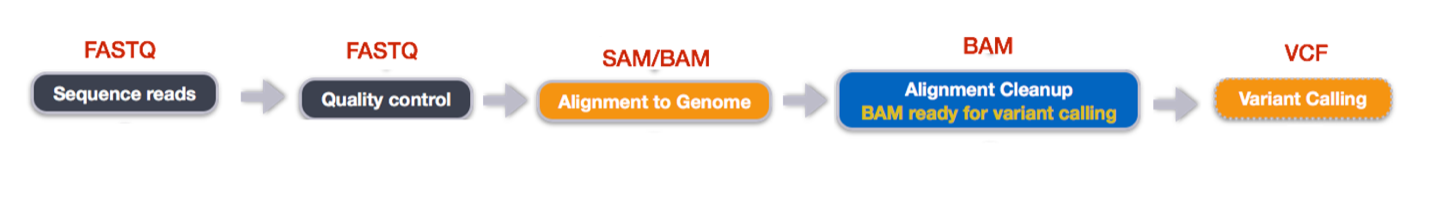
Once data is properly structured and stored, the next step is analysis. The workshop covered essential steps in RNA-seq analysis:
Sequencing Reads: RNA-seq begins with extracting total RNA from biological samples. The RNA is converted into complementary DNA (cDNA) via reverse transcription since sequencing technologies operate on DNA. The cDNA is then fragmented, ligated with adapters, amplified, and sequenced using high-throughput sequencing technologies like Illumina, PacBio, or Oxford Nanopore. This process generates raw sequence data representing the RNA transcriptome. The goal is to capture a comprehensive snapshot of gene expression levels across the entire transcriptome.
Quality Control (QC): Raw sequence data can be noisy, and often contains low-quality reads, contaminants, and technical artifacts. Quality control (QC) ensures data reliability using tools like FastQC, which assess:
- Base quality scores – accuracy of base calls.
- Adapter contamination – removal of sequencing
- GC content – detection of unexpected GC bias, which can indicate technical issues or problems with library preparation.
- Read length distribution – verification of expected read lengths.
- Overrepresented sequences – identification of contaminants or biases.
Alignment to the Genome: After quality control, the next step is aligning the cleaned reads to a reference genome. This process uses specialized algorithms and alignment tools like STAR, HISAT2, or Bowtie2. These tools map the short RNA-seq reads to known positions in the genome, allowing researchers to determine which genes are expressed and at what levels. The alignment process includes:
- Mapping reads while accounting for splicing events (since eukaryotic genes often contain exons and introns).
- Assigning reads to genes or transcript isoforms.
Alignment Cleanup: Post-alignment processing minimizes biases and improves accuracy. The cleanup step includes:
- Duplicate removal – tools like Picard or SAMtools can identify and eliminate PCR duplicates.
- Handling split reads – ensuring correct alignment of exon-exon junctions
- Refining the alignments – tools like GATK’s RealignerTargetCreator and BaseRecalibrator can be used to recalibrate base quality scores and realign reads around indels (insertions or deletions) to improve the accuracy of downstream variant calling.
Variant Calling: NA-seq data can reveal genetic variants such as SNPs (single nucleotide polymorphisms) and indels (insertions and deletions). Since RNA-seq reflects the transcriptome, variant calling in RNA-seq can also provide insight into alternative splicing events or mutations that affect gene expression. Key steps in this phase include:
- Variant identification – tools like GATK, FreeBayes, or Samtools mpileup are used to detect SNPs and indels by comparing the aligned reads to the reference genome. These tools consider read depth, allele balance, and base quality to call variants.
- Filtering variants based on quality score, read depth, and variant allele frequency
- Variant annotation using tools like ANNOVAR or VEP (Variant Effect Predictor). This step adds biological context to the variants, helping to determine their potential functional impact on genes, such as whether a variant is synonymous (no effect on protein) or nonsynonymous (affecting protein function).
Final Remarks
The Rutgers iJOBS genomics workshop provided an invaluable opportunity to develop practical skills in bioinformatics. Understanding the principles of data organization, sequencing workflows, and computational analyses is essential for modern biomedical research. As bioinformatics continues to drive scientific discovery, integrating biological knowledge with computational expertise will be key to unlocking the complexities of the genome.
This article was edited by Junior Editor E. Beyza Guven and Senior Editor Joycelyn Radeny.